
新智元报道
编辑:乔杨
虽然 AlphaFold 等系列的大模型已经在蛋白质预测方面取得了前所未有的突破,但依旧无法胜任蛋白质-蛋白质相互作用(PPI)这种复杂的任务。初创公司A-Alpha Bio 的 PPI 数据集 AlphaSeq,有望补足这方面的技术短板。
随着最近 AlphaFold 3 和 ESM 3 的相继推出,我们看到了深度学习在生物学领域的无限潜力。
然而,Dyno Therapeutics 的高级机器学习工程师 Abihishaike Mahajan 在上个月发布的一篇博文中指出了潜在的增长危机。

他认为,AlphaFold 系列所取得的成果,即将一个强大的深度学习模型应用于一个已经存在大量数据的领域,从而引发一场彻底的革命——这是极难复制的。
原因还是数据。我们几乎用尽了所有预先存在的数据,未经训练的蛋白质结构和序列正在枯竭,RNA 和 DNA 也是如此。
要想进一步训练模型,发掘更多来源和模态的数据是必不可少的。Mahajan 指出,理想情况下,这样的数据应该满足 3 个条件:
- 具有复杂的潜在分布
- 与重要的生理现象高度相关
- 适合大规模收集
在生物学领域,有很多数据可以满足前两个要求,比如蛋白形式测序、空间转录组学、体内测量和蛋白质-蛋白质相互作用等,但这类数据似乎很难大量采集、生成,形成规模化的数据集。
可喜的是,初创公司A-Alpha Bio 最近做出了这方面的突破。
他们最近发布的 AlphaSeq 数据库专注于蛋白质-蛋白质相互作用(protein-protein interaction, PPI),包含了超过 7.5 亿条测量结果,构成了世界上最大的 PPI 数据集。
在 AlphaSeq 数据的基础上,训练出的 AlphaBind 模型可以准确预测有不同结合特性(亲和力、特异性、交叉反应性、表位等)的蛋白质序列,从而辅助蛋白质设计或发现全新的蛋白质。

此外,作为实验平台,AlphaSeq 还能够同时定量测量数百万个 PPI 的结合亲和力,并快速得出结果,完美满足了规模化扩展的需求。
根据 CTO Randolph Lopez 的说法,他们目前每月执行约 30 次 AlphaSeq 检测,每次可以得到 100k~5M 个交叉点。这意味着,AlphaSeq 数据库还在以每月 3M~50M 的速度快速扩展。
A-Alpha Bio 这家初创公司也是大有来头。不仅有计算生物学领域的大牛 David Baker 作为科学顾问,联合创始人之一 David Younger 也是 Baker 实验室的校友。

David Baker 是华盛顿大学教授、蛋白质研究所所长。他领导团队从头开发的 Rosetta 算法奠定了用深度学习方法预测蛋白质结构的基础,揭开了 AlphaFold 和 ESMFold 的帷幕。
A-Alpha Bio 成立于 2017 年,根据 CrunchBase 的数据,他们已经融资 64.1M 美元,旨在通过合成生物学和机器学习技术来测量、发现、预测和设计蛋白质-蛋白质相互作用,从而加速药物开发的进程。
补足 AlphaFold
提到蛋白质相关的预测,你估计会疑惑:AlphaFold 还不够强大吗,为什么还需要开发新的数据和模型?
很遗憾,AlphaFold 的确不够强大,因为要了解蛋白质的相互作用(PPI)是一个相当复杂且困难的任务。
比如,要预测含有 13 个氨基酸的多肽与受体的结合效果,需要十多个不同的种子反复运行 AlphaFold,以及 MSA 子采样和其他一系列「技巧」,模型才能给出「某种程度上」正确的结构。
这个任务之所以如此复杂,主要源于 PPI 的复杂性。即使规定了蛋白质间作用力的空间,可能的结构数量也会随氨基酸数量呈指数级增长。
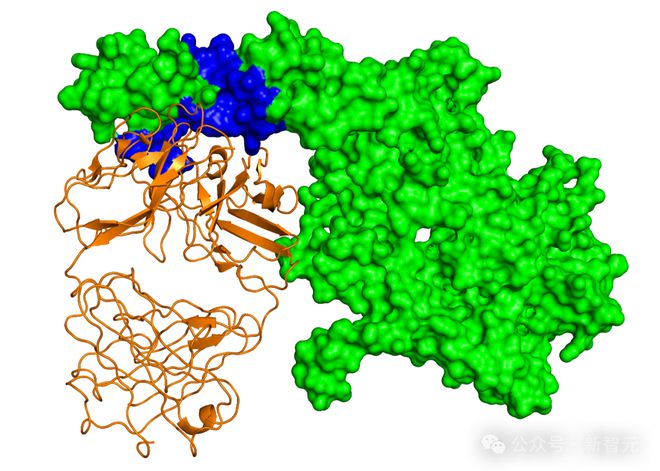
其中,分子构象的灵活性会导致不可预测的结合模式,并且潜在的相互作用表面的组合数量也会爆炸。
如果有足够的训练数据,模型也许能逐渐增强预测能力,应对问题的复杂性。
然而,传统的 PPI 数据规模相当有限,比如今年 1 月刚刚发布的 PDBbind+ 数据集,总共只包含 3176 个蛋白质-蛋白质复合物,远远无法满足生产级的蛋白质设计需求。

AlphaSeq 所用的方法,起源于 Baker 实验室在 2017 年发表的一篇论文,描述了A-Alpha Bio 对 PPI 数据进行大规模收集和表征的基本方法。

论文地址:https://www.pnas.org/doi/10.1073/pnas.1705867114#sec-1
酵母细胞立大功
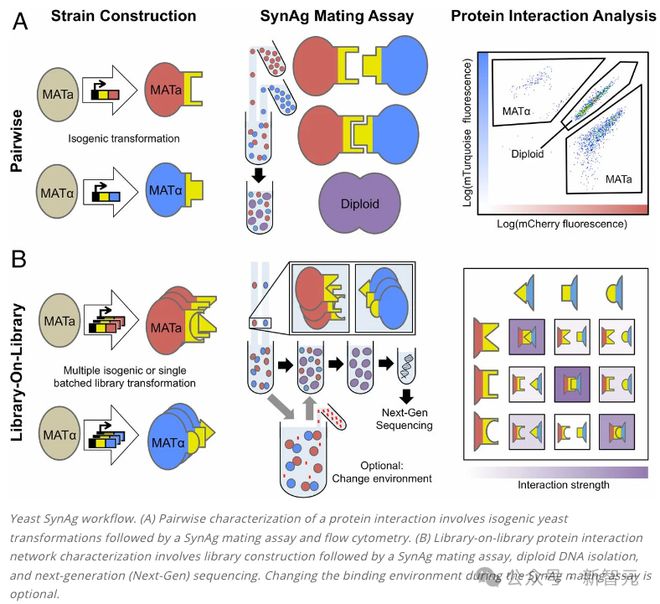
出乎意料的是,AlphaSeq 的原理是利用了酵母细胞的配对过程。
酵母细胞由两种类型的配子:MATa 和 MATα,它们在自然界中能够寻找到彼此并融合成为二倍体细胞。
这个过程就是由 MATa 细胞上的 Aga2 蛋白和 MATα细胞上的 Sag1 蛋白所介导的。当这些蛋白质相互作用时,它们会导致细胞粘在一起,促进配对并形成二倍体细胞。
AlphaSeq 正是利用了这个自然过程。研究人员对酵母细胞进行基因改造,让相关的蛋白质暴露在细胞表面,MATa 细胞搭载一组蛋白质,而 MATα细胞搭载另一组蛋白质。
将改造过的细胞进行混合时,它们配对的可能性就取决于表面蛋白质相互作用的强度。
那么如何快速测量数千万个蛋白质对之间的相互作用呢?答案是 DNA 编码库(DNA-encoded library)。
酵母细胞表面的每种蛋白质都与一个独特的「DNA 条形码」相关联。当两个酵母细胞配对时,这些条形码会在生成的二倍体细胞中聚集在一起。
通过一些基因工程的操作,这些 DNA 条形码最终会位于同一条染色体上的相邻位置。
在此基础上,我们就可以提取细胞 DNA 进行测序,两个 DNA 条形码相邻的频率就与两种蛋白质相互作用的强度直接相关。
值得注意的是,将整个平台都建立在酵母细胞上,可能存在根本限制。虽然酵母细胞表达的蛋白质和人体内的蛋白质之间具有高度可翻译性,但两者的翻译后修饰依旧存在差异。
翻译后修饰的差别可能会影响蛋白质的折叠,从而影响结合。
目前我们尚不清楚A-Alpha Bio 如何将收集的数据从酵母迁移到人类细胞,但他们已经对一些蛋白质的可翻译性进行了验证。这种方法至少总体上是可行且有效的。
应用前景
遗憾的是,A-Alpha Bio 目前还没有发布 AlphaSeq 的最新论文,关于 AlphaBind 模型的信息也十分有限。
但根据 Mahajan 文章的分析,该公司一系列产品有相当的应用前景。
对疾病治疗领域而言,可以帮助设计免疫细胞因子等药物;与大型制药公司合作,也可以帮助「分子胶」的开发。

使用 AlphaSeq 平台进行细胞因子亲和力调整来生成靶向免疫肿瘤治疗药物
参考资料:
https://www.owlposting.com/p/creating-the-largest-protein-protein
https://www.owlposting.com/p/wet-lab-innovations-will-lead-the






